[give_form id="30"]
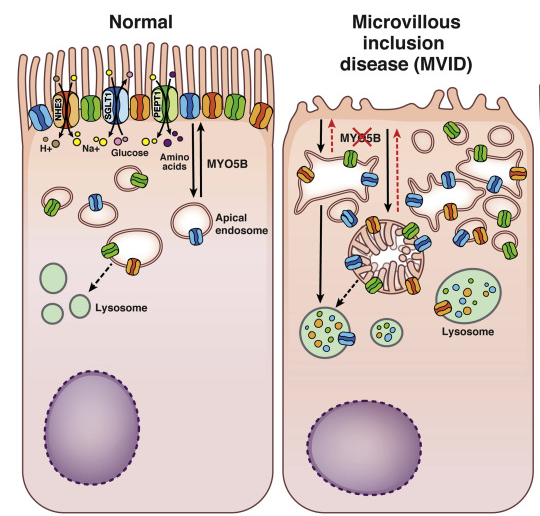
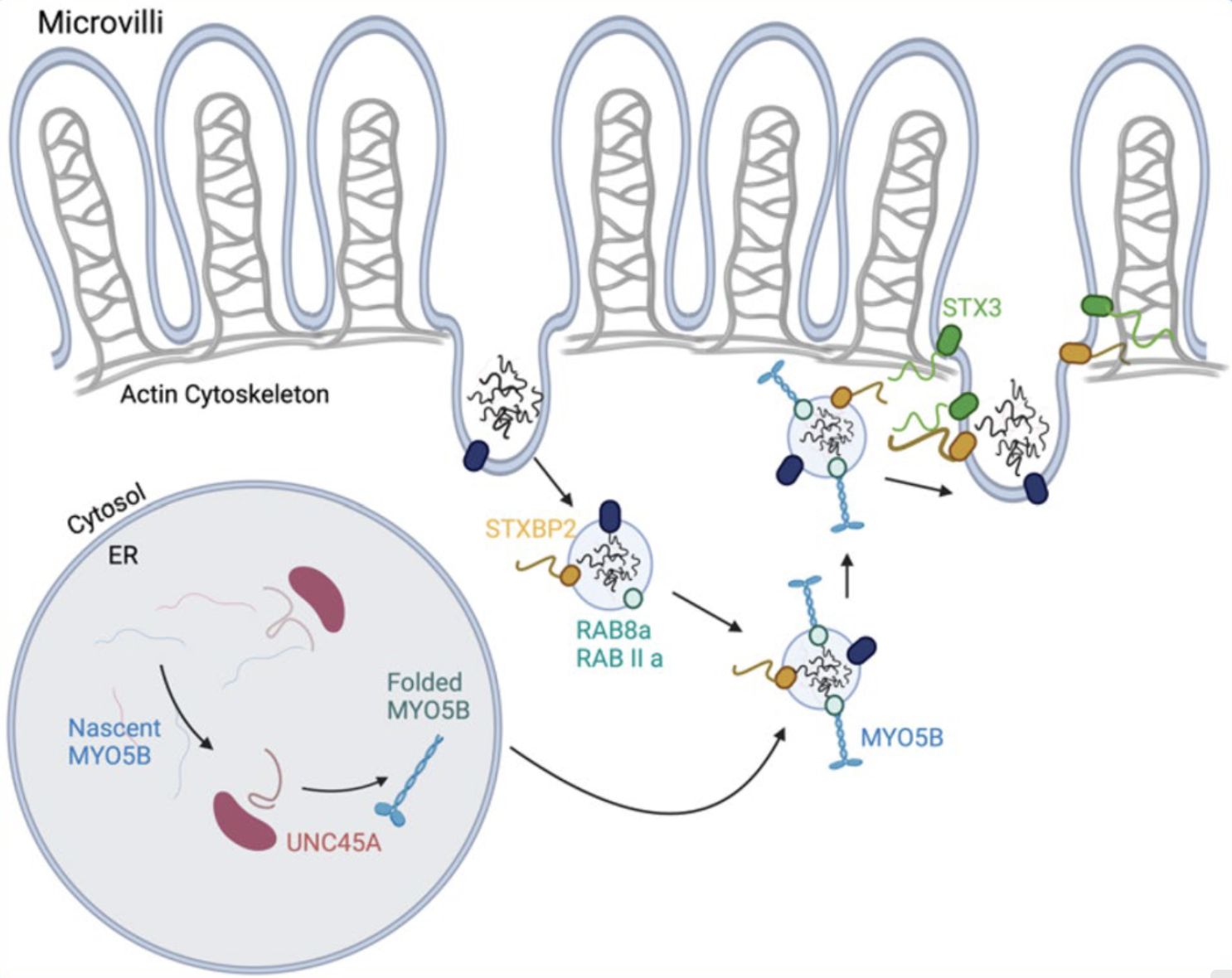
Die Mikrozotteneinschlusskrankheit (Microvillosity Inclusion Disease)
MVID



Angegliederter Verein
