[give_form id="30"]
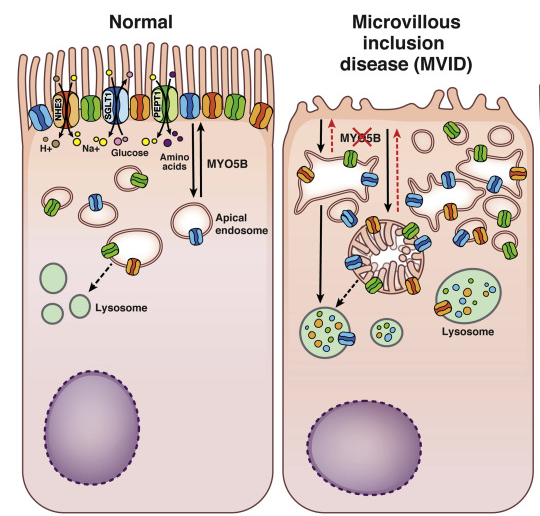
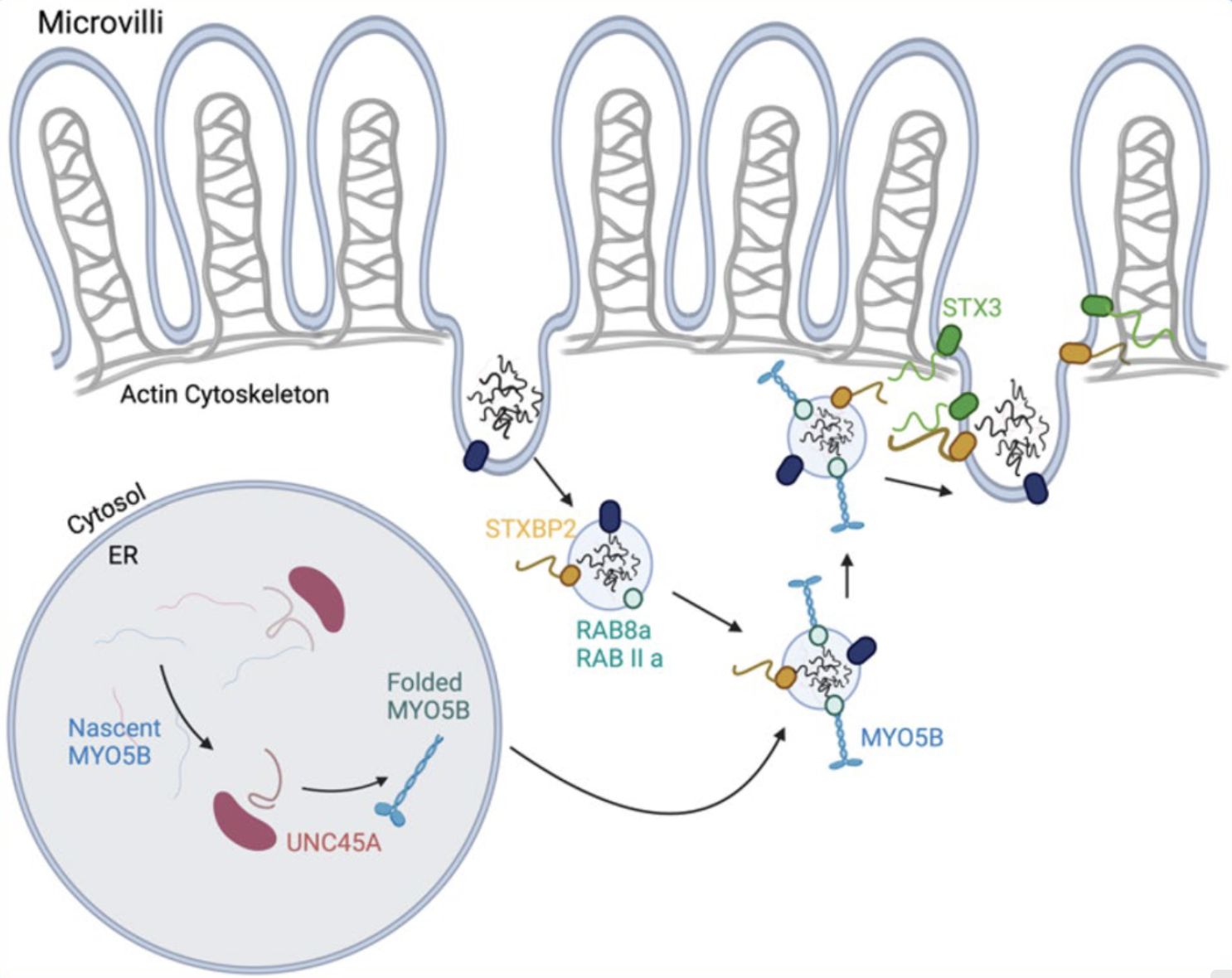
Enfermedad de inclusión microvellositaria
MVID



Asociación afiliada
