[give_form id="30"]
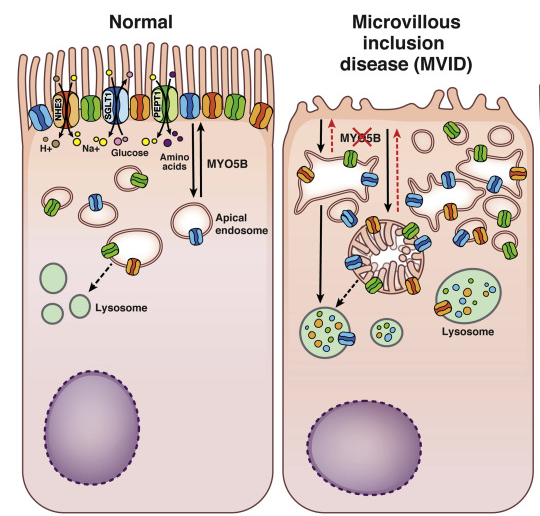
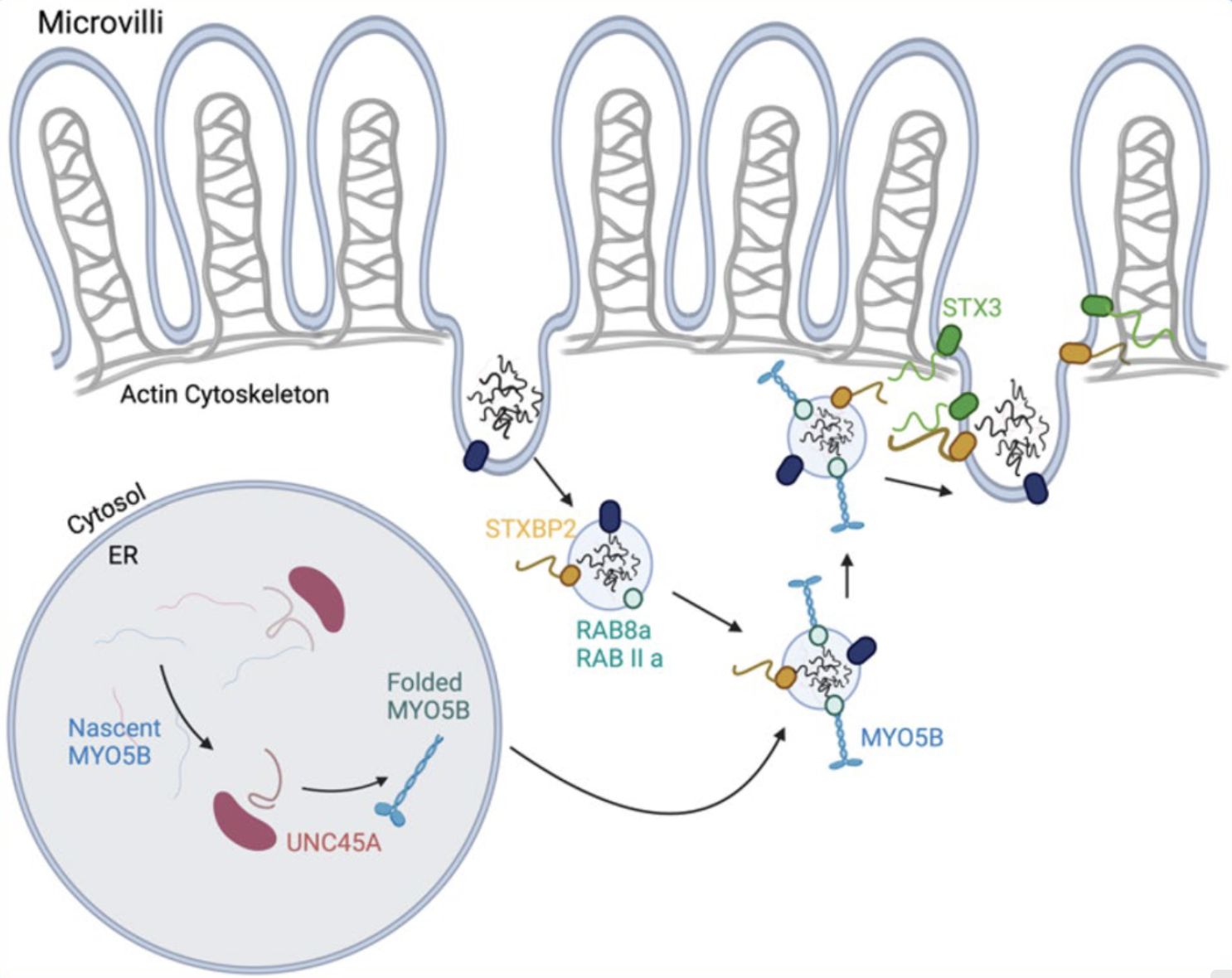
La Maladie des Inclusions Microvillositaires
MVID



Association Affiliée
